
This is a very recent Twitter thread from Josh Walkos concerning the testing of vaccines that are listed within the US CDC schedule of Childhood vaccines.
The question speaks for itself, as it addresses the safety analyses of each and every vaccine under that scheme. Normally an RCT trial will have a Trial Group and a Control (The “C “in RCT) group. The trial group are given the vaccine under trial and the control group get a placebo in order to compare any differences in reactions. For this to work properly the placebo needs to have nothing in it that can cause a reaction…especially one like an increase in antibodies.
Some of these below didn’t even have a trial group, and none of them used a genuine placebo according to the manufacturers evidence included in the packaging.
Tony Broomfield. Sen Editor.
Dangerous Globe
Mega Thread
The Childhood Vaccine Placebo Myth
Question – Is a true saline placebo used with the vaccine trials on the CDC childhood schedule?
All evidence I’ve provided is directly from the manufacturers insert or safety reports.
This is not my opinion so take it up with the FDA & Pharma if you’re mad about it.
1 – Kinrix
(Diphtheria-Tetanus-Acellular Pertussis-Hep B-Polio) – In the only trial specifically described in the package insert the control group received the Infanrix and polio vaccines. The package insert doesn’t mention any trial involving a placebo control group.
Following extracted from This Doc :- https://drive.google.com/file/d/1Ulz5HRP4ROFm49kQniiuqQ2vsRIFNH61/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates
observed in the clinical trials of a vaccine cannot be directly compared with rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
A total of 4,013 children were vaccinated with a single dose of KINRIX in 4 clinical trials. Of these, 381 children received a non-U.S. formulation of KINRIX (containing ≤2.5 mg 2-phenoxyethanol per dose as preservative).
The primary study (Study 048), conducted in the United States, was a randomized, controlled clinical trial in which children aged 4 to 6 years were vaccinated with KINRIX (n = 3,156) or control vaccines (INFANRIX and IPOL vaccine [IPV, Sanofi Pasteur SA]; n = 1,053) as a fifth DTaP vaccine dose following 4 doses of INFANRIX and as a fourth IPV dose following 3 doses of IPOL. Subjects also received the second dose of U.S.-licensed measles, mumps, and rubella (MMR) vaccine (Merck & Co., Inc.) administered concomitantly, at separate sites.
Data on adverse events were collected by parents/guardians using standardized forms for 4-5 consecutive days following vaccination with KINRIX or control vaccines (i.e., day of vaccination and the next 3 days). The reported frequencies of solicited local reactions and general adverse reactions in Study 048 are presented in Table 1.
In 3 studies (Studies 046, 047, and 048), children were monitored for unsolicited adverse events, including serious adverse events that occurred in the 31-day period following vaccination, and in 2 studies (Studies 047 and 048), parents/guardians were actively queried about changes in the child’s health status, including the occurrence of serious adverse events, through 6 months post-vaccination.
2 – Pediarix
(Diphtheria-Tetanus-Acellular Pertussis-Hep.B- Polio)
Control groups in the phase III trials received either the Infanrix vaccine along with hepatitis B, Hib, and polio vaccines, or other, unspecified vaccines. No control group received a placebo.
3 – Infanrix –
(Diphtheria-Tetanus-Acellular Pertussis) – Tested against a control group that received the DTP vaccine or no control group.
Following extracted from This Doc :- https://drive.google.com/file/d/1fUUkPH8gHd5fiBFhyZhGBl56fwLtmcCf/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. There is the possibility that broad use of INFANRIX could reveal adverse reactions not observed in clinical trials.
Approximately 95,000 doses of INFANRIX have been administered in clinical studies. In these studies, 29,243 infants have received INFANRIX in primary series studies, 6,081 children have received a fourth consecutive dose of INFANRIX, 1,764 children have received a fifth consecutive dose of INFANRIX, and 559 children have received a dose of INFANRIX following 3 doses of PEDIARIX.
Solicited Adverse Events: In a US study, 335 infants received INFANRIX, ENGERIX-B® [Hepatitis B Vaccine (Recombinant)], inactivated poliovirus vaccine (IPV, Sanofi Pasteur SA), Haemophilus b (Hib) conjugate vaccine (Wyeth Pharmaceuticals Inc.), and pneumococcal 7-valent conjugate (PCV7) vaccine (Wyeth Pharmaceuticals Inc.) concomitantly at separate sites. All vaccines were administered at 2, 4, and 6 months of age. Data on solicited local reactions and general adverse events were collected by parents using standardized diary cards for 4 consecutive days following each vaccine dose (i.e., day of vaccination and the next 3 days) (Table 1). Among subjects, 69% were White, 16% were Hispanic, 8% were Black, 4% were Asian, and 2% were of other racial/ethnic groups.
4 – DTP
(Diphtheria-Tetanus-Pertussis) – The vaccine was developed in the 1930s and has never been tested in an RCT against a control group receiving a real placebo.
Following extracted from This book :-  Available here
Available here
https://www.ncbi.nlm.nih.gov/books/NBK234363/pdf/Bookshelf_NBK234363.pdf

5 – Pentacel
Pentacel (Diphtheria-Tetanus-Acellular Pertussis-Polio-Hib) – The control groups in 3 of the 4 trials received an assortment of different vaccines. The 4th trial’s control group may have received no vaccines; however, its safety data is not presented in the package insert.
Following extracted from This Doc :- https://drive.google.com/file/d/1SB8zUchU9xp_j0eQTHent-znyta_oHec/view
6.1 Data from Clinical Studies
Rates of adverse reactions varied by dose number. The most frequent (>50% of participants) systemic reactions following any dose were fussiness/irritability and inconsolable crying. The most frequent (>30% of participants) injection site reactions following any dose were tenderness and increased circumference of the injected arm.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. The adverse reaction that appear to be related to vaccine use and for approximating rates of those events.
The safety of Pentacel was evaluated in four clinical studies in which a total of 5,980 participants received at least one dose of Pentacel. In three of the studies, conducted in the US, a total of 4,198 participants were enrolled to receive four consecutive doses of Pentacel. In the fourth study, conducted in Canada, 1,782 participants previously vaccinated with three doses of Pentacel received a fourth dose. The vaccination schedules of Pentacel, Control vaccines, and
concomitantly administered vaccines used in these studies are provided in Table 1.
Across the four studies, 50.8% of participants were female. Among participants in the three US studies, 64.5% were Caucasian, 9.2% were Black, 12.9% were Hispanic, 3.9% were Asian, and 9.5% were of other racial/ethnic groups. In the two controlled studies, the racial/ethnic distribution of participants who received Pentacel and Control vaccines was similar. In the Canadian fourth dose study, 86.0% of participants were Caucasian, 1.9% were Black, 0.8% were Hispanic, 4.3% were Asian, 2.0% were East Indian, 0.5% were Native Indian, and 4.5% were of other racial/ethnic groups.
6 – Daptacel
(Diphtheria-Tetanus-Acellular Pertussis) – Tested against a control group that received the DTP vaccine or no control group. In any event no inert placebo. (DG Note, Searching the document revealed 10 responses to the word Placebo but all 10 point to the use of other vaccines for compartison. Searching it again for “(S) saline” returned nothing)
Following extracted from This Doc :- There are 111 pages to this report which is highly fragmented to say the least https://drive.google.com/file/d/1CFrePXwN-q5ywCnuflnwLjUwScsLPvBU/view
Design: The study was double blinded, randomized, DT controlled trial. The original trial design was to include the U.S. whole cell vaccine manufactured by Wyeth-Lederle.
However, this vaccine was not available at the time the trial was to be initiated. The study was started as a three-arm trial (CPDT, DTaP-2, DT). Two months later the DTPwc vaccine was added as a fourth arm.
Separate randomization series were used for the three-armed and four-armed parts of the study.
Initial recruitment was to one of three vaccine: CPDT, DTaP-2 or DT control from March 1992 to April 1992, then to all four vaccines, including the DTPwc vaccine from May 1992 to February 1993.
Schedule: 2, 4 and 6 months
(note: No arm of the 3 and 4 armed trials was a saline placebo)
7 – Quadracel
(Diphtheria-tetanus – the control group in the insert does not mention an RCT with a saline placebo, only that it was tested against other vaccines like IPOL or MMR)
Following extracted from This Doc:- https://drive.google.com/file/d/1qIjY0SVED2Q8WxXhJj8DAXDJ725F6NVa/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to vaccine use and for approximating rates of those events.
In a randomized, controlled, multi-center study conducted in the US and Puerto Rico (Study M5I02; ClinicalTrials.gov Identifier: NCT01346293), 3372 children, 4 to 6 years of age, who had received 4 doses of DAPTACEL and/or Pentacel vaccine(s) received Quadracel, or DAPTACEL + IPOL (Poliovirus Vaccine Inactivated) vaccines administered concomitantly, but at separate sites.
Subjects also received Measles, Mumps, and Rubella Virus Vaccine Live (MMR) (Merck & Co., Inc.) and Varicella Virus Vaccine Live (Varicella vaccine) (Merck & Co., Inc.) administered concomitantly at separate sites. Safety was evaluated in 2733 subjects who received Quadracel and 621 subjects who received DAPTACEL + IPOL vaccines.
Among these subjects, 51.5% were male, 48.5% were female, 75.7% were Caucasian, 8.6% 82 were Black, 7.9% were Hispanic, 0.9% were Asian, and 7.8% were of other racial/ethnic groups. The mean age for both groups was 4.4 years and the ratio of male to female subjects and ethnicity were balanced between both groups.
Solicited injection site reactions and systemic reactions were collected daily for 7 days following vaccination, via diary cards. Participants were monitored for unsolicited adverse events for 28 days and serious adverse events (SAEs) for 6 months after vaccination. Solicited Adverse Reactions The incidence and severity of solicited injection site and systemic adverse reactions that occurred within 7 days after vaccination in each study group are shown in Table 1.
8 – Prevnar
Tested against a control group that received an experimental meningococcal vaccine, which at the time of use had not been approved.
Following extracted from This Doc: – https://drive.google.com/file/d/1VLR6NluMGK0E4yXUZM18IpUpi_MI7-MP/view
Results of Clinical Evaluations
Efficacy Against Invasive Disease
Efficacy was assessed in a randomized, double-blinded clinical trial in a multi-ethnic population at Northern California Kaiser Permanente (NCKP) from October 1995 through August 20, 1998, in which 37,816 infants were randomized to receive either Prevnar® or a control vaccine (an investigational meningococcal group C conjugate vaccine [MnCC]) at 2, 4, 6, and 12-15 months of age.
Prevnar® was administered to 18,906 children and the control vaccine to 18,910 children. Routinely recommended vaccines were also administered which changed during the trial to reflect changing AAP and Advisory Committee on Immunization Practices (ACIP) recommendations. A planned interim analysis was performed upon accrual of 17 cases of invasive disease due to vaccine-type S. pneumoniae (August 1998). Ancillary endpoints for evaluation of efficacy against pneumococcal disease were also assessed in this trial.
Invasive disease was defined as isolation and identification of S. pneumoniae from normally sterile body sites in children presenting with an acute illness consistent with pneumococcal disease.
Weekly surveillance of listings of cultures from the NCKP Regional Microbiology database was conducted to assure ascertainment of all cases. The primary endpoint was efficacy against invasive pneumococcal disease due to vaccine serotypes. The per protocol analysis of the primary endpoint included cases which occurred ≥14 days after the third dose. The intent-to-treat (ITT) analysis included all cases of invasive pneumococcal disease due to vaccine serotypes in children who received at least one dose of vaccine. Secondary analyses of efficacy against all invasive pneumococcal disease, regardless of serotype, were also performed according to these same per protocol and ITT definitions. Results of these analyses are presented in Table 1.
9 – Prevnar-13
Tested against a control group receiving Prevnar (older-generation vaccine).
Following extracted from This Doc: – https://drive.google.com/file/d/1_iI7Np-BfDmUwkQzuJWp46rkSBZ-ow5i/view
Clinical Trials Experience with Prevnar 13 in Adults ≥18 Years of Age
The safety of Prevnar 13 was assessed in 7 clinical studies (Studies 6-12) 6-12 conducted in the US and Europe which included 91,593 adults (48,806 received Prevnar 13) ranging in age from 18 through 101 years.
The 48,806 Prevnar 13 recipients included 899 adults who were aged 18 through 49 years, 2,616 adults who were aged 50 through 64 years, 45,291 adults aged 65 years and older. Of the 48,806 Prevnar 13 recipients, 46,890 adults had not previously received PPSV23 (“PPSV23 unvaccinated”) and 1,916 adults were previously vaccinated (“PPSV23 previously vaccinated”) with PPSV23 at least 3 years prior to enrolment.
10 – Hepatitis B: Engerix
Its side effect rate was compared to that of a previous generation product (plasma vaccine).
Following extracted from This Doc: – https://drive.google.com/file/d/1aZ1MtPiO58lE6Pjg0Ee_PZZ10c4iLjUs/view
Clinical Trials Experience
Based on clinical trial symptom sheet data, the incidence of local side effects is 24% and of systemic side effects 8%; both local and systemic side effects occurred in approximately 13% of subjects. The incidence of local and systemic reactions was comparable to those of plasma derived hepatitis B vaccines.
In a comparative trial in subjects from 11 years up to and including 15 years of age, the incidence of local and general solicited symptoms reported after a two-dose regimen of ENGERIX-B 20 μg was overall similar to that reported after the standard three-dose regimen of ENGERIX-B 10 μg. The safety profile presented below is based on data from more than 5,300 subjects.
11 – Twinrix
Tested in clinical trials against a control group that received separate hepatitis A and B vaccines.
Following extracted from This Doc: – https://drive.google.com/file/d/1K0vRj8CXuYtdhYUys4EPj2cG_niylk0I/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared with rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
As with any vaccine, there is the possibility that broad use of TWINRIX could reveal adverse events not observed in clinical trials. Following any dose of TWINRIX, the most common (≥10%) solicited injection site reactions were injection site soreness (35% to 41%) and redness (8% to 11%); the most common solicited systemic adverse events were headache (13% to 22%) and fatigue (11% to 14%).
The safety of TWINRIX has been evaluated in clinical trials involving the administration of approximately 7,500 doses to more than 2,500 individuals. In a US study, 773 subjects (aged 18 to 70 years) were randomized 1:1 to receive TWINRIX (0-, 1-, and 6-month schedule) or concurrent administration of ENGERIX-B (0-, 1-, and 6-month schedule) and HAVRIX (0- and 6-month schedule). Solicited local adverse reactions and systemic adverse events were recorded by parents/guardians on diary cards for 4 days (Days 0 to 3) after vaccination. Unsolicited adverse events were recorded for 31 days after vaccination.
Solicited events reported following the administration of TWINRIX or ENGERIX-B and HAVRIX are presented in Table 1.
12 – Recombivax HB
The package insert does not mention any safety RCT performed in infants.
Following extracted from This Doc: – https://drive.google.com/file/d/1LHJU_WAhXqewxvZJwWpRCRT7f4pHFaPk/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
In three clinical studies, 434 doses of RECOMBIVAX HB, 5 mcg, were administered to 147 healthy infants and children (up to 10 years of age) who were monitored for 5 days after each dose. Injection site reactions and systemic adverse reactions were reported following 0.2% and 10.4% of the injections, respectively. The most frequently reported systemic adverse reactions (>1% injections), in decreasing order of frequency, were irritability, fever (≥101°F oral equivalent), diarrhoea, fatigue/weakness, diminished appetite, and rhinitis. In a study that compared the three-dose regimen (5 mcg) with the two-dose regimen (10 mcg) of RECOMBIVAX HB in adolescents, the overall frequency of adverse reactions was generally similar .In a group of studies, 3258 doses of RECOMBIVAX HB, 10 mcg, were administered to 1252 healthy adults who were monitored for 5 days after each dose. Injection site reactions and systemic adverse reactions were reported following 17% and 15% of the injections, respectively.
13 – Hepatitis A – Havrix
The control group in the main trial received the hepatitis B vaccine. In three other trials, the control group received several other vaccines (MMR, varicella vaccine, and more).
Following extracted from This Doc: – https://drive.google.com/file/d/1XGppC-tPGSWvEZNGK8kRY15nGz76lxSA/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. The safety of HAVRIX has been evaluated in 61 clinical trials involving approximately 37,000 individuals receiving doses of 360 EL.U. (n = 21,928 in 3- or 4-dose schedule), 720 EL.U. (n = 12,274 in 2- or 3-dose schedule), or 1440 EL.U. (n = 2,782 in 2- or 3-dose schedule).
Coadministration Studies of HAVRIX in Children 11 to 25 Months of Age
In 4 studies, 3,152 children 11 to 25 months of age received at least one dose of HAVRIX 720 EL.U. administered alone or concomitantly with other routine childhood vaccinations [see Clinical Studies (14.2, 14.5)]. The studies included HAV 210 (N = 1,084), HAV 232 (N = 394), HAV 220 (N = 433), and HAV 231 (N = 1,241).
In the largest of these studies (HAV 231) conducted in the US, 1,241 children 15 months of age were randomized to receive: Group 1) HAVRIX alone; Group 2) HAVRIX concomitantly with measles, mumps, and rubella (MMR) vaccine (manufactured by Merck and Co.) and varicella vaccine (manufactured by Merck and Co.); or Group 3) MMR and varicella vaccines. Subjects in Group 3 who received MMR and varicella vaccines received the first dose of HAVRIX 42 days later. A second dose of HAVRIX was administered to all subjects 6 to 9 months after the first dose of HAVRIX.
14 – Vaqta – Hepatitis A
In one trial, there was no control group (according to another document, the control group received a compound that included aluminium and thimerosal), and in the second trial the vaccine was given concurrently with other vaccines and without a control group or by comparison with older study control groups.
Following extracted from This Doc: – https://drive.google.com/file/d/1LuPKwCve8Pguo-GJOzbOm1b9Hgu0Zn15/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
The safety of VAQTA has been evaluated in over 10,000 subjects 1 year to 85 years of age. Subjects were given one or two doses of the vaccine. The second (booster dose) was given 6 months or more after the first dose.
The most common local adverse reactions and systemic adverse events (≥15%) reported in different clinical trials across different age groups when VAQTA was administered alone or concomitantly were:
Children — 12 through 23 months of age: injection-site pain/tenderness (37.0%), injection-site erythema (21.2%), fever (16.4% when administered alone, and 27.0% when administered concomitantly).
Children/Adolescents — 2 through 18 years of age: injection-site pain (18.7%)
Adults — 19 years of age and older: injection-site pain, tenderness, or soreness (67.0%), injection-site warmth (18.2%) and headache (16.1%)
Allergic Reactions
Local and/or systemic allergic reactions that occurred in <1% of over 10,000 children/adolescents or adults in clinical trials regardless of causality included: injection-site pruritus and/or rash; bronchial constriction; asthma; wheezing; edema/swelling; rash; generalized erythema; urticaria; pruritus; eye irritation/itching; dermatitis [see Contraindications (4) and Warnings and Precautions (5.1)].
Children — 12 through 23 Months of Age
Across five clinical trials, 4374 children 12 to 23 months of age received one or two 25U doses of VAQTA, including 3885 children who received 2 doses of VAQTA and 1250 children who received VAQTA concomitantly with one or more other vaccines, including Measles, Mumps, and Rubella Virus Vaccine, Live (M-M-R II1), Varicella Vaccine, Live (VARIVAX1), Diphtheria and Tetanus Toxoids and Acellular Pertussis Vaccine, Adsorbed (Tripedia3 or INFANRIX2 ), Measles, Mumps, Rubella, and Varicella Vaccine, Live (ProQuad1), Pneumococcal 7-valent Conjugate Vaccine (Diphtheria CRM197 , Prevnar4), or Haemophilus B Conjugate Vaccine (Meningococcal Protein Conjugate, PedvaxHIB1 ).
Overall, the race distribution of study subjects was as follows: 64.7% Caucasian; 15.7% Hispanic American; 12.3% Black; 4.8% other; 1.4% Asian; and 1.1% Native American. The distribution of subjects by gender was 51.8% male and 48.2% female.
In an open-label clinical trial, 653 children 12 to 23 months of age were randomized to receive a first dose of VAQTA with ProQuad and Prevnar concomitantly (N=330) or a first dose of ProQuad and pneumococcal 7-valent conjugate vaccine concomitantly, followed by a first dose of VAQTA 6 weeks later (N=323). Approximately 6 months later, subjects received either the second doses of ProQuad and VAQTA concomitantly or the second doses of ProQuad and VAQTA separately. The race distribution of the study subjects was as follows: 60.3% Caucasian; 21.6% African American; 9.5% Hispanic-American; 7.2% other; 1.1% Asian; and 0.3% Native American. The distribution of subjects by gender was 50.7% male and 49.3% female.
15 ProQuad
Measles, Mumps, Rubella, Varicella (Chickenpox)
Safety was tested in several randomized clinical trials, most of which were not blinded. None of the trials contained a control group receiving only a placebo.
Following extracted from This Doc: – https://drive.google.com/file/d/13MxSgUKzQwZ59M2YZ_9Hwtc_2l7tSQVh/view
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. Vaccine-related adverse reactions reported during clinical trials were assessed by the study investigators to be possibly, probably, or definitely vaccine-related and are summarized below.
Children 12 Through 23 Months of Age Who Received a Single Dose of ProQuad Frozen ProQuad or refrigerator stable ProQuad was administered to 6038 children 12 through 23 months of age involved in clinical trials without concomitant administration with other vaccines. The safety of frozen ProQuad (N=4497) was compared with the safety of M-M-R II and VARIVAX given concomitantly (N=2038) at separate injection sites. The safety profile for ProQuad was similar to the component vaccines. Children in these studies were monitored for up to 42 days postvaccination using vaccination report card-aided surveillance. Safety follow-up was obtained for 98% of children in each group. Few subjects (<0.1%) who received ProQuad discontinued the study due to an adverse reaction.
The race distribution of the study subjects across these studies following a first dose of ProQuad was as follows: 67.2% White; 12.0% African American; 10.6% Hispanic; 5.0% Asian/Pacific; 3.4% other; 1.0% multiracial; 0.2% American Indian; 0.2% European; 0.2% Indian; and 0.1% Polynesian. The racial distribution of the control group was similar to that of the group who received ProQuad. The gender distribution across the studies following a first dose of ProQuad was 51.8% male and 48.2% female. The gender distribution of the control group was similar to that of the group who received ProQuad.
16 – MMR II
(Measles, Mumps, Rubella)
Tested in eight small unblinded clinical trials. All of the trials had one or more control groups receiving either the predecessor MMR vaccine, a measles-rubella (MR) vaccine, or a single-dose of the rubella vaccine.
Following extracted from This Doc: – https://drive.google.com/file/d/1IFm340mDs4z_GUMRASgVUWK8mzQnNpXx/view
A Merck document with little information on Trials
And this, a 215 page FOI Document detailing the 8 small trials. https://drive.google.com/file/d/1GKahQSNG8LvCAnEG7SGNyYPUEiSJwfd8/view
There is no evidence that trials used an inert placebo/saline solution, and a word search of the 237 page document
17 – MMR
(Measles, Mumps, Rubella) – Tested in several small to medium unblinded and partially randomized trials. The control groups totalled about 1/10 the number of subjects in the trial groups and received no injection.
Here is the product licensing report which is 237 pages long that has the studies referenced. https://drive.google.com/file/d/16qovZioEkWxDF739XeUdwvAyRk7unWm5/view
Quoted in the report is a Trial study document on MMR vaccine trials conducted in 1969. (See Page 12 of Referenced doc)
18 – Varivax
(Varicella) aka ChickenPox, – In one RCT the “placebo” given to the control group was actually the test vaccine from which the viral component was removed. Another trial compared two different formulations of the vaccine.
Following extracted from This Doc: – https://drive.google.com/file/d/13MxSgUKzQwZ59M2YZ_9Hwtc_2l7tSQVh/view
Which we also saw in the ProQuad 4 in #14 paper above
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice. Vaccine-related adverse reactions reported during clinical trials were assessed by the study investigators to be possibly, probably, or definitely vaccine-related and are summarized below.
Children 12 Through 23 Months of Age Who Received a Single Dose of ProQuad Frozen ProQuad or refrigerator stable ProQuad was administered to 6038 children 12 through 23 months of age involved in clinical trials without concomitant administration with other vaccines. The safety of frozen ProQuad (N=4497) was compared with the safety of M-M-R II and VARIVAX given concomitantly (N=2038) at separate injection sites. The safety profile for ProQuad was similar to the component vaccines. Children in these studies were monitored for up to 42 days postvaccination using vaccination report card-aided surveillance. Safety follow-up was obtained for 98% of children in each group. Few subjects (<0.1%) who received ProQuad discontinued the study due to an adverse reaction.
The race distribution of the study subjects across these studies following a first dose of ProQuad was as follows: 67.2% White; 12.0% African American; 10.6% Hispanic; 5.0% Asian/Pacific; 3.4% other; 1.0% multiracial; 0.2% American Indian; 0.2% European; 0.2% Indian; and 0.1% Polynesian. The racial distribution of the control group was similar to that of the group who received ProQuad. The gender distribution across the studies following a first dose of ProQuad was 51.8% male and 48.2% female. The gender distribution of the control group was similar to that of the group who received ProQuad.
The only vaccine-related injection-site adverse reaction that was more frequent among recipients of ProQuad than recipients of M-M-R II and VARIVAX was rash at the injection site (2.4% versus 1.6%, respectively, risk difference 0.9%, 95% CI: 0.1, 1.5).
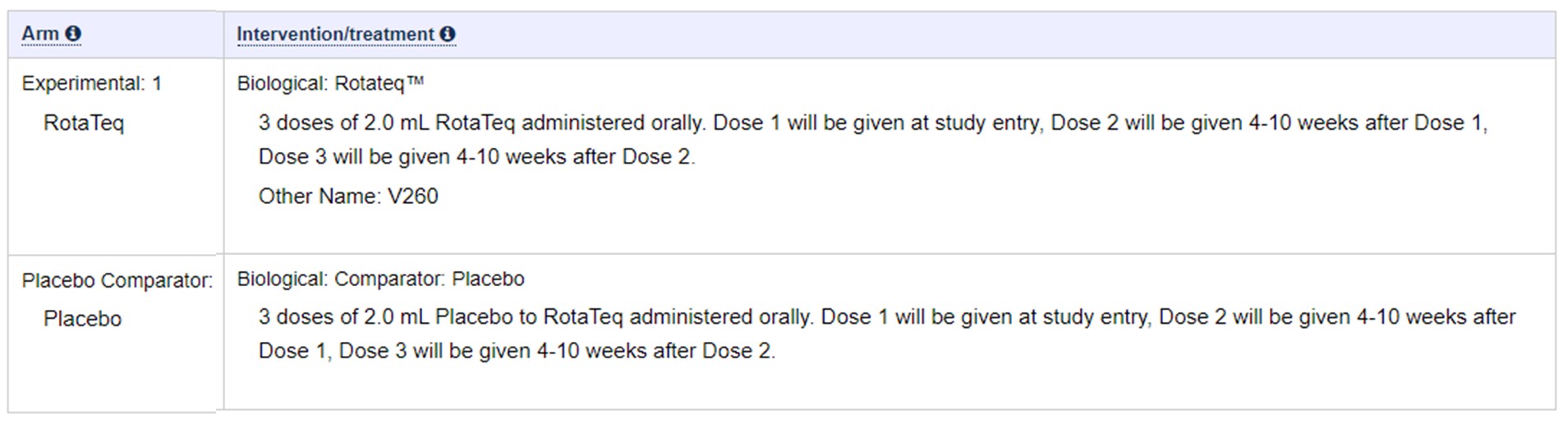
19 – Rotavirus- RotaTeq
Following extracted from This Doc: – https://clinicaltrials.gov/ct2/show/NCT00090233?term=rotavirus
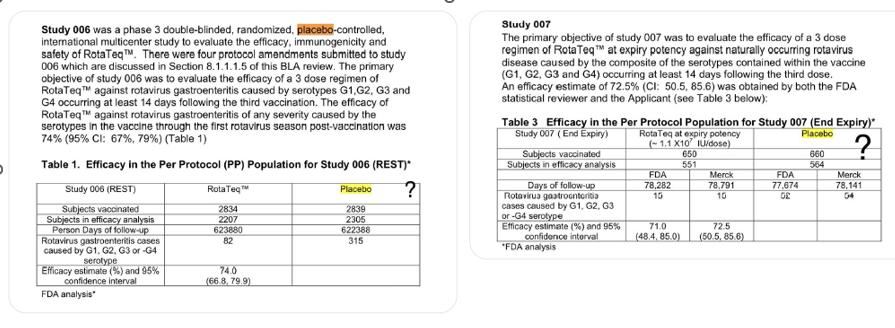
20 – Rotarix
The control group in the trial received the vaccine-sans-antigen compound.
Following extracted from This Doc: – https://drive.google.com/file/d/1T5ZQZYINtJhdrYGHrwSZ1M5HWFKf4dK8/view
“I know there are a lot of zealots out there that will invariably attack me for this but I have this to say to you. I have provided links to direct source documents from the FDA and manufacturers and even screenshots to make it easier for you.
If you disagree, I challenge you to find me one Phase 3 clinical trial used for approval on the current Childhood Vaccine Schedule that uses a true placebo, an inert SALINE PLACEBO, not another vaccine or bio-active placebo.
It must be from source documents used in the trials that were used for the basis for approval. Again, I’ve only provided what the FDA and Manufacturers submitted, this isn’t opinion or conjecture.
This isn’t an “anti-vax” thing, it’s an examination of how safety is determined for children and if you truly cared about that you would be calling for True Placebos to be used which are much cheaper to use anyway.
So, the question is why would a pharmaceutical company not use a saline placebo unless they are obscuring safety signals? Furthermore, why would our regulatory agencies allow this obvious tactic? #placebo
If you’ve found this thread informative, please consider giving me a follow to stay up to date with future threads. Thanks for reading. Josh 🙏.
- • •
Champagne Joshi @JoshWalkos ·
Follow Here is a list of all of my threads so far for ease of access. I appreciate the support, more to come. Thread Topics
- COVID-19 Vaccines
- VAERS
- The PCR “Test”
- Masks
- Lockdowns
- mRNA Approval for Kids
- Post Autopsies
- Excess Death
11:25 PM · Jan 14, 2023 Read the full conversation on Twitter 895 See the latest COVID-19 information on Twitter Read 22 replies
Comment from the Dangerous Globe
The idea that using vaccines in place of placebos is startling and scary. Some of the very old types of vaccines never had any safety testing, and now as each generation goes through testing it is compared with the last gen technology, based on the previous model which was based on nothing at all but thin air.
Also noticeable is the use in one trial of a vaccine that had not even been accepted by the authorities at the time of use.
Some of the Childrens vaccine trials use previous versions that have had their active ingredients removed. The rationale for using any kind of vaccine in place of a Saline solution is that there ‘may be a chance of an illness during the trial that may be staved off using a form of vaccine rather than an inert compound.
How in blue blazes does a vaccine that has no active ingredient stave off anything? If anything, the child who gets this placebo shot runs the risk of a modified vaccine (with all that it contains) with no single possibility of benefit.
Just to put the tin hat on it , running a supposedly RCT trial with no control group at all?
(and they passed Peer review and FDA/CDC scrutiny as well)
Words fail us….


